

Wel of niet opereren bij ernstige verstopping?
Veel kinderen in Nederland hebben last van verstopping (obstipatie). Vaak gaat dit vanzelf over. Ook leefstijladviezen in combinatie met een laxeermiddel kunnen helpen. Een kleine groep kinderen blijft, ondanks het gebruik van medicijnen, last houden van ernstige klachten zoals pijnlijke, harde en vaak grote ontlasting, het niet kunnen ophouden van ontlasting en buikpijn. Verstopping die niet met medicijnen is op te lossen noemen we therapieresistente obstipatie (TRO). Bij een klein deel van deze patiënten is een operatie misschien de oplossing van de problemen, maar het is niet duidelijk welke kinderen dit écht kan helpen.

De ziekte van Kawasaki: een opgelost raadsel
De ziekte van Kawasaki is een zeldzame aandoening die bij 1 op de 10.000 kinderen in Nederland voorkomt en lijkt op andere infectieziekten omdat de klachten hetzelfde zijn. De ziekte van Kawasaki zorgt bij jonge kinderen voor ontsteking van de bloedvaten, vooral die rond het hart. Er is nog geen bloedtest om de ziekte van Kawasaki snel en nauwkeurig vast te stellen. Een snelle behandeling is noodzakelijk om schade aan het hart te voorkomen. In het Amsterdam UMC werken onderzoekers aan het ontwikkelen van een bloedtest om de ziekte van Kawasaki op een goede en betrouwbare manier vast te stellen.

Eerdere diagnose en behandeling voor kinderen met CIDP
Chronische inflammatoire demyeliniserende polyradiculoneuropathie (CIDP) is een zeldzame ziekte van de zenuwen die spierverlamming, gevoelloosheid en tintelingen veroorzaakt. CIDP is bij kinderen nog zeldzamer dan bij volwassenen, waardoor de ziekte vaak laat wordt ontdekt. Een vroege behandeling van CIDP vergroot de kans op herstel. Omdat CIDP zo weinig voorkomt bij kinderen, wordt soms een verkeerde diagnose gesteld en een verkeerde behandeling gestart.

Belangrijke gezondheidswinst bij XLA door vroege opsporing en geoptimaliseerde behandeling
Hielprikscreening voor X-gebonden agammaglobulinemie
“Als het maar gezond is”, zeggen aanstaande ouders vaak. Maar wat als je kind altijd ziek is en de ene oorontsteking nog niet voorbij en de volgende koortsperiode al weer aanbreekt? Voor ouders van kinderen met de ernstige afweerstoornis XLA (X-gebonden agammaglobulinemie) is dit vaak werkelijkheid. XLA-Patiënten hebben een genetische defect in het BTK-gen, waardoor de B-celontwikkeling in het beenmerg verstoord is en zij geen B-cellen hebben. B-cellen zijn witte bloedcellen die antistoffen produceren. Door het gebrek aan antistoffen krijgen alle XLA-patiënten ernstige infecties. Als zij niet worden behandeld, overlijden zij aan hun ziekte. Heel soms wordt XLA bij de eerste infecte al vastgesteld, maar in de meeste gevallen gaan families door een onzekere, verdrietige periode, met ernstige infecties en chronische gezondheidsschade, veel ziekenhuisbezoeken en prikmomenten bij hun jonge kind. Er is behoefte aan een vroege opsporing van XLA om zo snel mogelijk te starten met de behandeling. Het opnemen van XLA in de hielprikscreening bij pasgeborenen is een belangrijke stap.
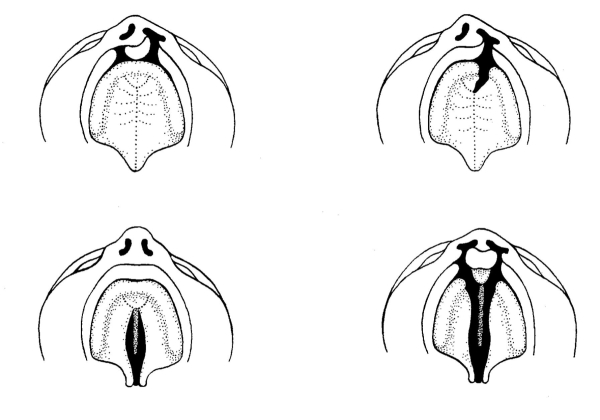
Betere spraak bij kinderen met een schisis na gehemeltesluiting
Onderzoek naar de operatietechniek met de beste spraakresultaten en de minste complicaties
Schisis is de medische term voor een gespleten lip, kaak en/of gehemelte. Het is een zeldzame aangeboren afwijking die in Nederland per jaar bij ongeveer 300 kinderen wordt vastgesteld. Schisis wordt multidisciplinair behandeld in zeven centra in Nederland, waaronder het Wilhelmina Kinderziekenhuis (WKZ) in Utrecht. Kinderen met een schisis hebben onder andere voedings-, gehoor- en spraakproblemen. Voor het eerste levensjaar worden vaak al een of twee operaties gedaan om de lip en/of het gehemelte te sluiten. In het WKZ blijken per jaar ongeveer 30 kinderen na een operatie slecht verstaanbaar zijn vanwege een zogenaamde ‘open neusspraak’. Vele ziekenhuisbezoeken, intensieve logopedie en soms aanvullende operaties zijn nodig om dit te verbeteren, het is echter onduidelijk welke operatie de beste spraak oplevert.

Gezond gewicht en bewegen bij kinderen met zeldzame syndromen
Bij kinderen met het zeldzame genetische Angelman syndroom en Dup15q syndroom zijn door een verandering in het UBE3A-gen hun groei, gewicht en bewegingsmogelijkheden verstoord. Kinderen met het Angelman syndroom hebben vaak overgewicht en problemen met lopen, terwijl kinderen met het Dup15q syndroom juist ondergewicht hebben. Onderzoekers in het Erasmus MC / Sophia Kinderziekenhuis zoeken naar de samenhang tussen voeding, stofwisseling en beweging bij deze kinderen. Met deze inzichten kan de zorg en begeleiding worden verbeterd.
Wel of niet behandelen bij screening op CF?
Onduidelijke diagnose bij positief op CF gescreende baby’s
Cystic fibrosis (CF) of taaislijmziekte is een zeldzame erfelijke ziekte die ontstaat door een fout in het CFTR-gen. Dit gen maakt het CFTR-eiwit (chloorkanaal) aan dat zorgt voor goed, soepel slijm in veel organen en op veel plekken in het lichaam. Werkt dit niet goed, dan raakt de water- en zoutbalans in het lichaam verstoord. Dit leidt tot taaier slijm waardoor allerlei problemen in het lichaam ontstaan. Alle pasgeborenen in Nederland worden via de hielprik gescreend op CF. De 25-30 kinderen per jaar met een positieve uitkomst voor CF ondergaan een zogenaamde ‘zweettest’ om het zoutgehalte vast te stellen en daarmee CF te bevestigen. Bij 10 kinderen per jaar is de diagnose na deze test onduidelijk. Dit zorgt voor onzekerheid en angst bij de ouders over de gezondheid van hun kind en over het wel of niet nodig zijn van behandelingen. Meerdere jaren wordt bij deze groep vervolgonderzoek gedaan: een herhaling van de zweettest en afname van een darmbiopt om in verse darmcellen de activiteit van het CFTR-eiwit te meten. Er is grote behoefte aan sneller en minder belastend vervolgonderzoek voor deze groep.

Ernstige prematurenretinopathie voorspellen met behulp van kunstmatige intelligentie
Oogzorg op maat bij te vroeg geboren kinderen
Prematurenretinopathie (Retinopathy of Prematurity, ofwel ROP) is een zeldzame, ernstige oogaandoening bij te vroeg geboren kinderen. Deze kan ontstaan doordat de bloedvaten in het netvlies niet goed uitgroeien. Bij te late behandeling, kan ernstige ROP leiden tot levenslange beperkingen van het zicht of zelfs blindheid. Daarom is regelmatige en intensieve screening van te vroeg geboren kinderen door een oogarts tijdens de eerste levensmaanden belangrijk. De belastende oogscreenings blijken achteraf vaak onnodig, omdat de meeste gescreende kinderen geen ernstige ROP ontwikkelen. Het is op dit moment nog niet mogelijk om in een vroeg stadium een nauwkeurige inschatting te maken welke kinderen een verhoogd risico op ernstige ROP hebben.
Internationaal register MLC
Een beter beeld krijgen van de hersenziekte MLC
Megalencefale Leukoencefalopathie met subcorticale Cysten (MLC) is een zeldzame, erfelijke hersenziekte die zich kenmerkt door een chronische zwelling van het hersenweefsel. Doordat er te veel water in de witte stof van de hersenen zit, functioneert deze niet goed. Gevolgen zijn een abnormaal groot hoofd, motorische problemen, vertraging en later ook achteruitgaan van het denken, en epilepsie. MLC is (nog) niet te genezen. Wel begrijpen onderzoekers steeds beter hoe de ziekte ontstaat. Met deze kennis komt ook een behandeling in zicht.

Betere vooruitzichten voor kinderen met het GNAO1-syndroom
Het GNAO1 syndroom is een niet te genezen zeldzame, erfelijke ziekte waarbij sprake is van ernstige, soms levensbedreigende bewegingsstoornissen, een ontwikkelingsachterstand en vaak epilepsie. Kenmerk van de bewegingsstoornissen bij een deel van de kinderen met het GNAO1 syndroom is een plotselinge, onvoorspelbare verslechtering. Deze onvoorspelbaarheid heeft een enorme impact op de kwaliteit van leven van het kind en de ouders. Voor de bewegingsstoornissen is behandeling mogelijk, maar onduidelijk is welke behandeling voor welk kind het beste is.

Opereren van pasgeborenen met een zeldzame aangeboren cysteuze longafwijking
Onnodig of noodzaak?
Een aangeboren cysteuze longafwijking is een zeldzame longaandoening. 80-90% van de pasgeborenen met deze aandoening heeft geen klachten. Wereldwijd discussiëren artsen al meer dan 25 jaar over de beste behandeling voor deze groep pasgeborenen met een cysteuze longafwijking en zónder klachten. Sommige artsen pleiten voor niet opereren en regelmatige controles. Anderen geven de voorkeur aan een operatie waarbij het aangedane deel van de long wordt weggehaald. Zo kan de behandeling in verschillende ziekenhuizen binnen één land (ook binnen Nederland) verschillen. Voor ouders is het onduidelijk of een operatie noodzakelijk is als hun kind geen klachten heeft.

RELAX
Het verminderen van pijn en angst bij kinderen met sikkelcelziekte
Nederland telt ongeveer 1.000 kinderen en 800 volwassenen met sikkelcelziekte. Kinderen met sikkelcelziekte hebben vaak ernstige pijnaanvallen waarvoor zij in het ziekenhuis worden opgenomen. De behandeling met sterke pijnstillers die volgt geeft veel bijwerkingen. De pijnaanval en de ziekenhuisopname kunnen zó aangrijpend zijn, dat de stressreactie vaak langer dan 3 maanden aanhoudt. Dit noemen we Post Traumatische Stressstoornis (PTSS). Door de pijn en angst die de kinderen ervaren wordt de pijn nog sterker en komen zij in een negatieve spiraal terecht. De volgende pijnaanval is vaak nog ernstiger. De negatieve spiraal van angst en pijn waar kinderen met sikkelcelziekte in terecht kunnen komen kan helaas nog niet met een medische behandeling worden gestopt.

Go with the (lymphatic) flow
Verbeteren diagnose en behandeling bij kinderen met aangeboren (centrale) lymfevatafwijkingen
Bij kinderen met zeldzame aangeboren (centrale) lymfevataandoeningen (CCLA) zijn de grote lymfevaten in het lichaam niet goed aangelegd, hierdoor kan de afvoer van lymfevocht blokkeren of lymfevocht lekken. In lymfevocht zitten belangrijke afweerstoffen en eiwitten. Lekt er lymfevocht weg, dan ontstaat een verhoogd risico op infecties, stollings- en schildklierproblemen. Bij blokkering kan lymfevocht in de borst- en buikholte en/of onderhuids opstapelen wat bij pasgeborenen vaak zorgt voor ernstige ademhalingsproblemen. Het risico op overlijden van deze kinderen is groot. In Nederland worden per jaar gemiddeld 20 kinderen met CCLA geboren.

Ademnood en luchtwegproblemen voorkomen bij baby’s met een slokdarmafsluiting
Slokdarmafsluiting (oesofagusatresie) is een zeldzame, aangeboren afwijking die bij 1 op 4.100 pasgeborenen voorkomt. Omdat een deel van de slokdarm (de buis die de mond met de maag verbindt) ontbreekt, kunnen deze baby’s geen enkel voedsel opnemen. Zij worden met spoed opgenomen in het ziekenhuis, krijgen voeding via een infuus en worden snel geopereerd. De operatie zorgt dat de slokdarm weer wordt verbonden met de maag.

Kiezen voor de beste behandeling van slokdarmachalasie bij kinderen
Slokdarm-achalasie is een heel zeldzame ziekte die bij 5-10 kinderen in Nederland per jaar wordt vastgesteld. De spier aan de onderkant van de slokdarm ontspant niet meer, waardoor eten in de slokdarm vast komt te zitten en niet meer verder kan naar de maag. De kinderen hebben pijn bij het slikken en steeds meer klachten. Worden deze kinderen niet behandeld, dan is voedsel toedienen via een sonde de enige optie.

Oog voor de toekomst
Blindheid voorkomen bij patiënten met de ziekte van Hurler
De ziekte van Hurler is een zeer ernstige, zeldzame stofwisselingsziekte. In Nederland zijn er ongeveer 25 kinderen met deze ziekte. Zij bezoeken voor hun behandeling allemaal het Wilhelmina Kinderziekenhuis (WKZ) in Utrecht.
Vooral hersenen, skelet en ogen zijn aangedaan. Door stamceltransplantatie kan de ziekte in de hersenen worden gestopt. Onderzoekers willen weten of met stamceltransplantatie de ziekte in andere delen van het lichaam ook effectief wordt behandeld. Daarom komen de patiënten jaarlijks voor controle terug in het WKZ. Uit deze controles blijkt dat de ziekte in de ogen vaak niet is gestopt. Tieners die een succesvolle stamceltransplantatie ondergingen, zijn vaak nachtblind en slechtziend door achteruitgang van het netvlies. Een behandeling voor deze netvliesziekte is er niet.
Effectieve en minder belastende operatie om heup-uit-de-kom te voorkomen
Voor kinderen met cerebrale parese
De For Wis(h)dom Foundation levert voor drie jaar een bijdrage aan onderzoek waarbij wordt gekeken of een nieuwe, minder belastende operatie bij kinderen met cerebrale parese ervoor kan zorgen dat de heup in de kom blijft. Als het onderzoek laat zien dat deze nieuwe operatie effectief is dan wordt bij deze kinderen een grotere en belastende operatie op latere leeftijd voorkomen. Dit draagt bij aan een betere kwaliteit van leven van zowel de ouders als het kind zelf.

Galgangatresie
Plakjes galweg en galblaas als model om galwegatresie te onderzoeken
Galwegatresie is een zeldzame aandoening van de galwegen die in Nederland jaarlijks bij gemiddeld 10 pasgeborenen voorkomt. De galwegen van de lever naar de darm zijn verschrompeld, waardoor de gal niet meer kan doorstromen. Pasgeborenen met deze aandoening worden hierdoor erg ziek en krijgen geelzucht. Alle kinderen met deze aandoening worden behandeld in het expertisecentrum voor galwegatresie in het UMCG.

‘Kinderen zoals ik!’
Het dagelijks functioneren van kinderen met een complexe hersengerelateerde aandoening.
Kinderen met complexe hersengerelateerde aandoeningen en hun ouders bezoeken voor behandeling het Kinderhersencentrum van het Erasmus MC-Sophia kinderziekenhuis. Zij hebben bijvoorbeeld een erfelijke ontwikkelingsstoornis, een stoornis in de aanleg van de hersenen of het ruggenmerg of op jonge leeftijd opgelopen hersenschade. Vaak zijn de aandoeningen zeldzaam. Ondanks dat het verschillende aandoeningen zijn, hebben de kinderen vaak vergelijkbare problemen bij bijvoorbeeld het bewegen, denken of hun gedrag.Het Kinderhersencentrum wil deze kinderen graag de beste behandeling geven, ervoor zorgen dat ze optimaal kunnen meedoen en een goede kwaliteit van leven hebben. Het centrum volgt de ontwikkeling van deze kinderen op verschillende gebieden over de jaren heen. Kinderen en ouders vullen hiervoor af en toe vragenlijsten in. De behandelend artsen spreken de uitkomsten daarvan met de ouders en kinderen.

Wigschedel
Is een operatie bij kinderen met een wigschedel zinvol?
Als bij een pasgeboren baby de schedel niet normaal kan groeien, belemmert dat ook de groei van de hersenen. Een schedel kan alleen normaal groeien als de schedelnaden goed functioneren. Craniosynostose is een zeldzame aandoening waarbij één of meerdere schedelnaden al voor de geboorte zijn gesloten. Dit verstoort de groei van de hersenen. Als gevolg hiervan kan de hersendruk te hoog worden, waardoor het zicht van het kind en het leervermogen slechter worden. Kinderen met craniosynostose worden om deze reden aan hun schedel geopereerd binnen hun eerste levensjaar.
Syndrome4Life
Verbeteren van de medische zorg bij (jong)volwassenen met zeldzame genetische syndromen door internisten
Wij zijn verheugd om een project te ondersteunen dat zich richt op het verbeteren van de medische zorg bij (jong) volwassenen met een zeldzaam genetisch syndroom.
De komende jaren worden duizenden jongvolwassenen met een zeldzaam syndroom van de kinderarts overgedragen aan internisten. Deze hebben vaak onvoldoende kennis over syndromen en zien geen rol voor zichzelf bij de zorg voor deze groep omdat zij zich niet bewust zijn van de mogelijke orgaan- en hormoonproblemen bij deze patiënten. Verkeerde of onnodige behandelingen worden gestart en belangrijke problemen over het hoofd gezien.
Door het creëren van awareness en het tegelijkertijd beschikbaar maken van aanbevelingen voor internisten, worden gemiste diagnoses en verkeerde of onnodige behandelingen bij vele duizenden (jong)volwassen patiënten met een zeldzaam syndroom voorkomen.

PROM
Vragenlijst set (PROM-set) ontwikkelen voor patiënten met een zeldzaam genetisch syndroom
Ongeveer 3% van de Nederlanders heeft een zeldzame ziekte als gevolg van een genetisch syndroom met complexe lichamelijke, verstandelijke en psychiatrische problemen. Het ‘Emma Center for Personalized Medicine’ in het Amsterdam AMC richt zich op het verbeteren van de zorg en behandelingen voor deze groep. Het meten van de effectiviteit van deze behandelingen is een grote uitdaging. Goede meetinstrumenten ontbreken en de problemen verschillen per patiënt. Wanneer een behandeling geen effect lijkt te hebben is het de vraag of de behandeling onvoldoende is of dat het effect niet meetbaar is. Ook meten de meetinstrumenten vaak niet wat patiënten belangrijk vinden. Daarom ontwikkelen de onderzoekers een set vragenlijsten (PROM-set) die vaststelt hoe deze patiënten hun eigen gezondheid en kwaliteit van leven ervaren. De resultaten van deze ‘patient reported outcome measures’ (PROMs) zijn bruikbaar voor patiëntenzorg en wetenschappelijk onderzoek.
Vanishing White Matter
Medicijnen voor de wittestofziekte VWM
Wittestofziekte Vanishing White Matter (VWM) is een zeldzame, erfelijke hersenziekte waarbij de witte stof in de hersenen verdwijnt. De ziekte komt bij 1 op de 100.000 pasgeborenen voor.
Witte stof is het bedradingssysteem tussen de zenuwcellen in de hersenen. Zenuwcellen communiceren met elkaar en met het lichaam en sturen zo het lichaam en het denken aan. Door een fout in het EIF2b-gen wordt de witte stof aangetast. Het lichaam doet niet meer wat de zenuwcellen in de hersenen vragen. De gevolgen zijn enorm: patiënten raken ernstig lichamelijk beperkt en overlijden uiteindelijk. Er is geen behandeling voor de witte stofziekte.
For Wis(h)dom gaat voor een periode van 2 jaar een bijdrage leveren aan dit project.
Het dilemma van wel of niet opereren bij te vroeggeboren kinderen met ernstige NEC
Een keuzehulp ontwikkelen voor ouders en artsen met behulp van kunstmatige intelligentie
NEC is een vaak ernstig verlopende acute darmontsteking bij meestal (veel) te vroeg geboren kinderen. In Nederland gaat het jaarlijks om ongeveer 200 kinderen. Deze te vroeggeboren kinderen overleven steeds vaker en daardoor neemt het aantal kinderen met NEC toe. Rond de 40% van de kinderen met NEC wordt geopereerd. Bij de helft van deze groep is de operatie succesvol; de andere helft overlijdt. 70% van de kinderen die de operatie overleven heeft voor de rest van het leven last van lange-termijn complicaties van deze operatie. Ouders en artsen vragen zich steeds vaker of een operatie wel in het belang van het kind is. For Wis(h)dom ondersteunt dit onderzoek van 2022 tot en met 2024.

Expertise Connected
Expertisenetwerken voor zeldzame aandoeningen – samenwerken aan zorg en wetenschappelijk onderzoek
Het ministerie van VWS, de NFU (Nederlandse Federatie van Universitair Medische Centra) en de VSOP (Patiëntenkoepel voor zeldzame en genetische aandoeningen) spannen zich sinds enkele jaren in om inzicht te krijgen in welke ziekenhuizen kennis en ervaring op het gebied van zeldzame aandoeningen aanwezig is. Door VWS erkende ziekenhuizen mogen zich expertisecentrum noemen. In Nederland zijn er ongeveer 300 erkende expertisecentra voor zeldzame aandoeningen. Deze centra hebben een belangrijke rol bij wetenschappelijk onderzoek, maar geven patiënten met een zeldzame aandoening ook de best mogelijke behandeling en begeleiding.
